液相常见的压力问题,基线问题,峰形问题等都有解答,小伙伴们赶紧寻找答案吧。话不多说,直接上干货。何谓反相柱、正相柱?
答:“反相”和“正相”的概念是液相色谱法早期提出的概念,当时键合相色谱柱尚未出现,固定相被涂覆在载体表面,极易流失,为此科学家对流动相使用给出了合理的建议:流动相极性与固定液极性应具有较大差别,以减少固定液流失。固定相极性弱于流动相时的液相色谱法被称为反相色谱法,固定相极性强于流动相时的液相色谱法被称为正相色谱法。尽管目前键合相色谱柱已成为主流,但这一概念在色谱方法开发、预测出峰顺序等方面具有重要意义。
由上面的介绍可知具体的色谱方法、色谱柱属于正相还是反相不仅取决于固定相极性,同时还取决于流动相极性。C18(硅胶键合十八烷基硅烷)、C8(硅胶键合辛基硅烷)、PH(硅胶键合苯基硅烷)等色谱柱,由于固定相极性极低,比目前已知的任何流动相的极性都要低,因而是标准的反相柱;Silica(硅胶)、NH2(硅胶键合氨丙基硅烷)具有较高的极性,主要用于分离带有极性基团的化合物,所用流动相的极性通常低于这些固定相,因而是标准的正相柱。CN(硅胶键合腈丙基)的极性适中,当流动相极性超过CN时,它属于反相柱,反之则是正相柱。色谱柱规格对分析结果会产生何种影响?
答:色谱柱内径决定载样量,载样量与内径的平方成正比;色谱柱长度与塔板数成正比,与柱压成正比;粒径影响涡流扩散相,粒径越小涡流扩散相越小,柱效越高,粒径与柱效近似成反比;粒径越小,压力也越大,压力与粒径的平方成反比。填料孔径对分析对象的分子量有限制,当孔径为分析物尺寸的5倍以上时,分析物才能顺利通过孔隙,孔径处于60~120 Å的色谱柱适用于相对分子量小于10000的分析物,孔径为300 Å的色谱柱可以满足分子量处于10000以上的大分子化合物分析。液相色谱分析中如何才能提高分离度?答:下式为分离度计算公式
N:柱效(Efficiency)反映色谱柱性能,柱效越高,分离度越好。在其他条件恒定的情况下,塔板数增加一倍,分离度仅提高40%。操作中,可通过下面两种方式增加塔板数进而提高分离度:其一,使用长柱或双柱串联,但也会使分离时间大大延长;其二,使用细粒径填料的色谱柱,但这需要耐更高压力的液相色谱系统。相比之下后者更为可取。
α:选择性(selectivity)是指色谱柱-流动相体系分离两个化合物的能力。选择性主要与固定相、流动相组成以及柱温等因素有关,与保留值也密切相关,其中固定相和流动相组成影响较大。以最常见的反相模式为例,反相柱(包括C18、C8、PH等)是以分配作用对化合物进行保留的,不同化合物的分离是基于它们在键合相与流动相中分配系数的差异,如果两种化合物的水溶性、在烷烃-水体系的分配系数等方面存在明显差异,那么这些化合物通常是能够利用反相柱达到分离;PH柱对具有苯环的化合物具有特殊保留。
正相模式下,硅胶柱、胺基柱、氰基柱与带有极性基团的化合物之间存在极性相互作用,对化合物的基团具有选择性,常常用于结构类似物、异构体化合物的分离。流动相方面,降低流动相的洗脱强度通常可以增大分离度;而有机溶剂类型也会影响分离,比如反相条件下,乙腈和甲醇的选择性就存在很大差异,这种差异需要在实践中摸索,但无论如何,多种溶剂类型带给我们更多的实现分离的可能。
k:随着容量因子k的增大,分离度也随之增加,这种影响在k值较低时非常明显,当k值大于10时,k值增加对分离度的影响就不再显著,这就告诫无原则地提高k值以增大分离度是没有意义的。增加键合相密度能够提高k值;另外改变键合基团类型也能改变k值,比如在反相色谱中,随着键合相碳链长度的增加,k值逐渐增大。色谱峰的峰形是怎样衡量的?有何规定?理论上讲,色谱峰应符合高斯曲线分布,然而实际上任何色谱峰都对高斯曲线分布存在一定的偏离,亦即不对称。峰拖尾可以用不对称因子(As)或USP拖尾因子(Tf)来衡量,显然不对称因子的说法更准确,因为色谱峰存在前延、完美对称、拖尾三种形态。一般来说,制药行业以USP拖尾因子作为评测标准,而其他行业则多采用As来测定峰形。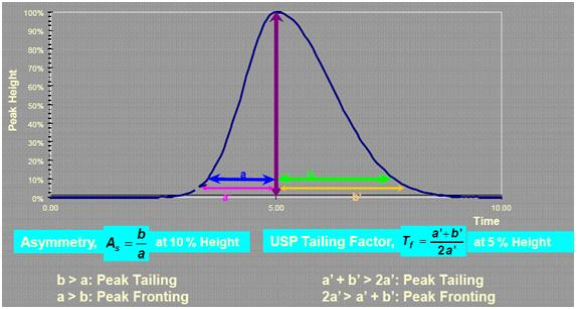
对于药物分析,通常有明确的规定,Tf应处于某一范围之间,比如我国药典规定某些药物的拖尾因子应处于0.95-1.05之间。其他行业尚无较为明确的规定。什么是梯度洗脱?什么情况下使用梯度洗脱?
答:为了改善分析结果,某些操作需要连续改变流动相中各溶剂组分的比例以连续改变流动相的极性,使每个分析组分都有合适的容量因子k,并使样品种的所有组分可在最短时间内实现最佳分离,这种洗脱方式称为梯度洗脱。
梯度洗脱可在下列情形中发挥重要作用:
A 在等度下具有较宽k值的多种样品分析。
B 大分子样品分析。
C 样品含有强保留的干扰物,在目标化合物出峰后设置梯度洗脱,将干扰物洗脱出来,以免其影响下一次数据采集。
D 分析方法建立时,不知道其洗脱情况,使用梯度洗脱,找出其较优的洗脱条件。何谓封端?封端的意义是什么?硅胶表面的硅羟基(Si-OH)密度为8 μmol/m2,由于空间位阻的存在,硅烷化键合反应最多只能覆盖50%的硅羟基,超过一半的硅羟基是活性硅羟基。这些硅羟基与化合物的极性基团存在极性相互作用和离子交换作用,为化合物的保留增加了不被期望的作用力,往往会影响峰形。用短链氯硅烷(如三甲基氯硅烷)键合活性的硅羟基,可以减小甚至消除这种影响,这种操作被称为封端(Endcap)。
封端处理的意义:
抑制了特异性吸附,提高了色谱峰的对称性,并改善了分离效果;
在一定程度上掩蔽了硅胶表面,使其对碱性环境的耐受性增强;
通过空间位阻掩盖了键合反应形成的Si-O-Si键,使其对酸性环境的耐受性增强;可能会影响对极性样品的选择性。柱床塌陷和键合相塌陷柱床塌陷是指色谱柱使用一段时间后色谱柱入口处的柱床产生可见的空隙。该空隙的存在增大了死体积,会导致色谱柱柱效下降。造成柱床塌陷的原因如下:
其一,色谱柱填装时的压力过低,填装不紧密,在高压下使用一段时间,开始出现空隙;
其二,操作压力超出色谱柱填料的耐压值,导致填料颗粒破碎产生空隙;
其三,流动相溶解填料导致空隙出现。键合相塌陷是指由于流动相极性与键合相极性相差太大,键合相无法在流动相中充分伸展而倒伏、缠结在一起,比如普通C18在纯水相条件下。相塌陷会导致色谱柱对化合物的保留不足。系统压力升高的原因及排除使用者通过观察仪器的系统监测掌握系统的压力,如果发现压力偏高,不要立即认为“柱压高了”,因为系统压力通常由柱前压力、色谱柱压力和流通池压力加合而成。此时正确的做法是:在操作条件下,测定系统压力,得到p总;卸下色谱柱,在相同条件下,测定柱前压力,得到p前;用两通将泵流出管路与流通池连接,在操作条件下,测定压力,得到p(前+后)。通过计算找出压力高是来自柱前、色谱柱还是柱后。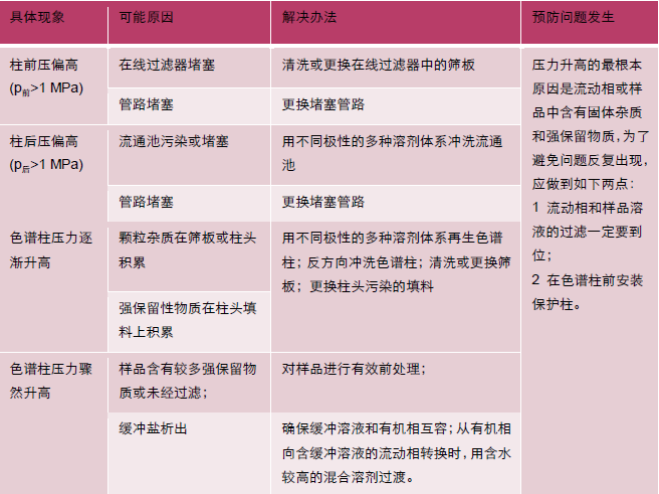
以上情形假设没有安装保护柱,如果安装了保护柱,压力升高时应先测试保护柱的压力,以便确认问题是否来自保护柱。系统压力不稳定通常情况下,泵压的变动值超过了0.5 Mpa,系统压力就属于不稳定的范畴。导致系统压力不稳定的最直接原因为流动相流量和组成输出的不稳定,而导致流动相输出不稳定的原因通常包括溶剂互容性较差、系统漏液、系统存在气泡以及输液系统部件工作失效。下面将介绍排除“系统压力不稳”的方法。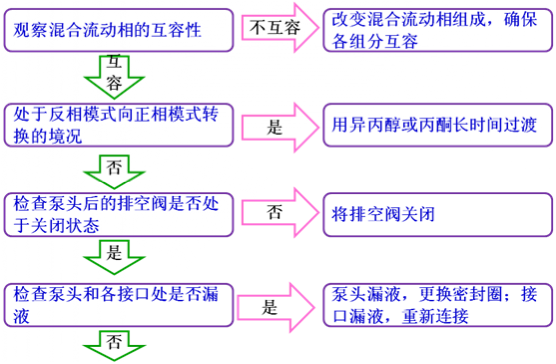
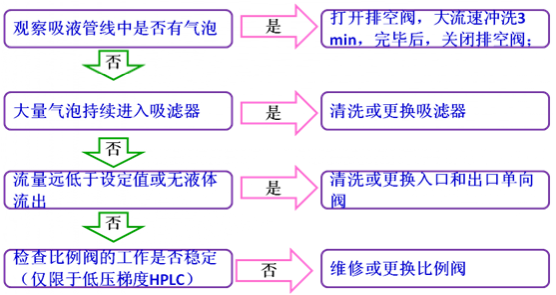
流动相输出不稳定还会导致基线不稳定以及保留时间变化等现象,所以基线不稳定或保留时间不重现时,先看一下柱压是否稳定,如果不稳定,三个问题可以合并处理。基线不稳定
基线不稳定是指色谱曲线在较长时间范围内对响应值的0刻度较大的偏离,通常包括基线漂移(单方向)、基线起伏(规律的上下起伏)、区域宽度极大的色谱峰三种情况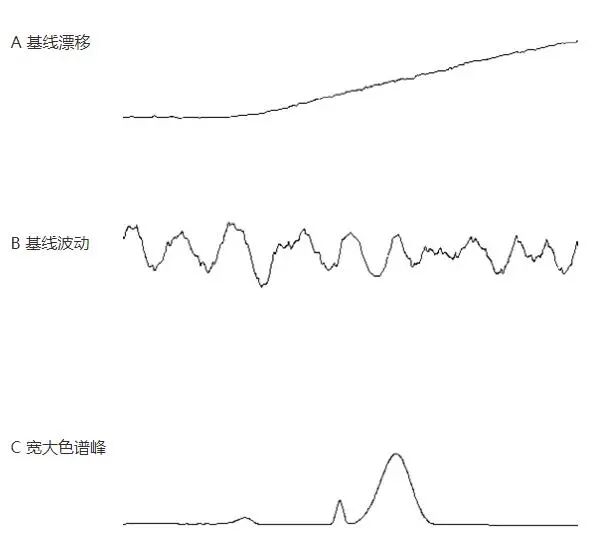
如果基线不稳定同时伴随压力波动,请按照“压力不稳定”进行排查。如果压力稳定,请按下表中的方法进行排查。
噪声升高
噪声(baseline noise):由检测器输出与被测样品组分无关的无规则波动信号,在特定灵敏度下用响应单位表示。可分为高频噪声和短周期噪声两种。对于光谱型检测器,噪声的正常响应值处于0.000005 V ~ 0.00002 V之间(参考Shimadzu 的HPLC仪器),高于此值,即可认为噪声偏高。
噪声升高时,先按下列步骤排查流动相问题
噪声升高时,若流动相没有问题,应排查仪器的问题
柱性能下降
柱性能下降的一个重要现象就是在保留时间基本不发生变化的情况下,色谱峰的区域宽度明显增大(如下图)。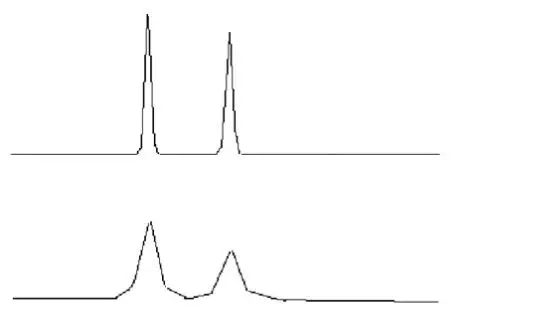
当遇到柱效下降,请按下表进行排查和解决
保留时间重现性差
当保留时间的偏差超过2%,可以认为保留时间的重现性差。对于某一个特定的分析,保留时间主要受流动相组成、流速、温度以及固定相影响,当保留时间发生明显变化时,应从这几方面入手。
A 由流动相造成的保留时间不稳定
B 由固定相造成的保留时间不稳定
峰形不理想理想的色谱峰遵循正态分布,应该符合高斯曲线,是完美而对称的峰形(如图A);然而由于化合物性质的特殊性、色谱柱故障、操作条件等问题存在,峰形可能会不对称,前延(B)或拖尾(C)。
峰形前延(B)
中药成分分析项目中,提取溶剂通常为乙醇、甲醇、乙酸乙酯等洗脱强度较强的溶剂,此时易发生“样品溶剂的洗脱强度远远超过流动相”的情况,为避免前延峰出现,可以用通过“流动相稀释提取液”或降低“进样体积”的方式。
峰形拖尾(C)
鬼峰鬼峰(Ghost peak):是对未知来源的色谱峰的统称。
气泡峰气泡峰是指携带气泡的流动相通过检测器时得到的响应值;这里的气泡可能是流动相中释放的空气,也可能是有机相遇水放热而会释放的有机溶剂分子;气泡峰往往具有较高的响应值,区域宽度仅为数秒,出峰时间和相应值比较有规律(如下图)。通常只有光谱型检测器才会出气泡峰。
解决办法:
脱气
使用多元梯度时,对每一种组分进行脱气,排除它们溶解的空气;
对于预先混合的溶剂,混合后脱气,排除溶解的空气和因放热而挥发出的有机溶剂分子;
前面的脱气方式对于预先混合好的混合溶剂,基本可以达到消除气泡峰的目的;但对于多种纯溶剂在仪器中混合的情况,混合后溶剂体系放热,存在潜在的气泡,当流动相流出色谱柱时,压强骤减,潜在的气泡溢出,被检测器检测到。为了防止这一问题出现,应在检测器出口连接一段内径较细的管路,使流动相通过检测器时,环境压强较高,气泡难以溢出,可以避免气泡峰出现。
 手机版
手机版




